
Kaikki iLive-sisältö tarkistetaan lääketieteellisesti tai se tarkistetaan tosiasiallisen tarkkuuden varmistamiseksi.
Meillä on tiukat hankintaohjeet ja vain linkki hyvämaineisiin mediasivustoihin, akateemisiin tutkimuslaitoksiin ja mahdollisuuksien mukaan lääketieteellisesti vertaisarvioituihin tutkimuksiin. Huomaa, että suluissa ([1], [2] jne.) Olevat numerot ovat napsautettavia linkkejä näihin tutkimuksiin.
Jos sinusta tuntuu, että jokin sisältö on virheellinen, vanhentunut tai muuten kyseenalainen, valitse se ja paina Ctrl + Enter.
Aivojen lissensefalia
Lääketieteen asiantuntija

Viimeksi tarkistettu: 12.07.2025
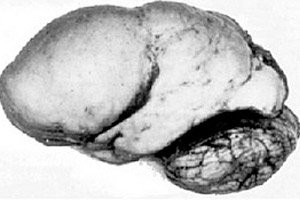
Orgaanisista aivopatologioista erottuu sellainen synnynnäinen aivojen kehityksen poikkeavuus kuin lissenkefalia, jonka ydin on sen aivopuoliskojen kuoren lähes sileä pinta – riittämätön määrä kierteitä ja vakoja. [ 1 ]
Jos konvoluutioita ei ole lainkaan, määritellään agyria, ja useiden leveiden, litteiden konvoluutioiden esiintymistä kutsutaan pachygyriaksi. Näillä vioilla, kuten joillakin muilla aivojen reduktiomuodonmuutoksilla, on ICD-10:ssä koodi Q04.3.
Epidemiologia
Harvinaisten sairauksien tilastojen mukaan lissenkefaliaa esiintyy 1–1,2 tapausta 100 000 vastasyntynyttä kohden. [ 2 ], [ 3 ]
Joidenkin tietojen mukaan jopa 25–30 % klassisen lissenkefalian tapauksista havaitaan Miller-Diekerin oireyhtymää sairastavilla lapsilla; LIS1- ja DCX-geenien pistemutaatioita ja deleetioita havaitaan lähes 85 %:lla potilaista. [ 4 ]
Lissenkefaliaan liittyvien 17 geenin geneettiset tutkimukset osoittivat, että LIS1-mutaatio tai -deleetio on syynä 40 %:lle potilaista ja 23 %:lla DCX-mutaatioon, jota seuraavat TUBA1A (5 %) ja DYNC1H1 (3 %).[ 5 ]
Syyt Lissensefalia
Kaikki tunnetut syyt aivokuoren (cortex cerebri) lähes tai kokonaan ilman poimuja ja uurteita, jotka lisäävät ihmisaivojen "työskentelyaluetta" ja varmistavat keskushermoston "tuottavuuden", liittyvät perinataalisen kehityksen häiriöihin. Toisin sanoen sikiöllä kehittyy lissenkefalia. [ 6 ]
Sikiön aivokuoren kerrosten muodostumisen epäonnistuminen lissenkefaliassa on seurausta sitä muodostavien hermosolujen epänormaalista migraatiosta tai tämän prosessin ennenaikaisesta lopettamisesta.
Tämä aivokuoren histogeneesille välttämätön prosessi tapahtuu useissa vaiheissa raskauden 7. ja 18. viikon välillä. Ja koska se on lisääntynyt herkkyydellään geneettisille mutaatioille sekä erilaisille negatiivisille fysikaalisille, kemiallisille ja biologisille vaikutuksille, mikä tahansa poikkeama normista voi johtaa hermosolujen virheelliseen lokalisaatioon ja mahdolliseen harmaan aineen paksuuntumiseen aivokuoren alueelle ilman sille ominaista rakennetta. [ 7 ]
Joissakin tapauksissa lasten lissenkefalia liittyy Miller-Diekerin, Walker-Warburgin tai Norman-Robertsin oireyhtymiin.
Lue myös – Aivojen kehityshäiriöt
Riskitekijät
Joidenkin geenien mutaatioiden lisäksi riskitekijöitä tällaisen vakavan vian omaavan lapsen syntymälle ovat sikiön hapenpuute (hypoksia); aivojen riittämätön verenkierto (hypoperfuusio); akuutti aivoverenkiertohäiriö perinataalisen aivohalvauksen muodossa; istukan patologiat; raskaana olevan naisen virusinfektiot (mukaan lukien TORCH); [ 8 ] yleisen aineenvaihdunnan ja kilpirauhasen toiminnan ongelmat; tupakointi, alkoholi, psykotrooppiset ja huumaavat aineet; useiden lääkkeiden käyttö; kohonneet säteilytasot. [ 9 ]
Synnyssä
Kaikissa lissenkefalian tapauksissa patogeneesi ei johdu kromosomipoikkeavuuksista ja geenimutaatioista. Mutta tunnetaan joitakin geenejä, jotka koodaavat proteiineja, joilla on tärkeä rooli neuroblastien ja hermosolujen oikeassa liikkumisessa säteittäisten gliasolujen pitkin – aivokuoren muodostamiseksi. Ja näiden geenien mutaatiot johtavat tähän patologiaan. [ 10 ]
Nämä ovat erityisesti satunnaisia mutaatioita (ilman perinnöllisyyttä) LIS1-geenissä kromosomissa 17, joka säätelee mikrotubulusten sytoplasmista motoriproteiinia dyneiiniä, sekä DCX-geenissä X-kromosomissa, joka koodaa doublekortiini-proteiinia (lissenkefaliini-X). [ 11 ]. Ensimmäisessä tapauksessa asiantuntijat määrittelevät klassisen lissenkefalian (tyyppi I), toisessa X-kromosomiin kytkeytyneen. [ 12 ]
Kun fosfoproteiini filamiini 1:tä koodaava FLN1-geeni poistetaan, suunnattu hermosolujen migraatio ei välttämättä ala ollenkaan, mikä johtaa konvoluutioiden (agyrian) täydelliseen puuttumiseen. [ 13 ]
CDK5-geenissä on tunnistettu mutaatioita. Kinaasientsyymi on solunsisäisen aineenvaihdunnan katalysaattori, joka säätelee keskushermoston hermosolujen solusykliä ja varmistaa niiden normaalin migraation aivorakenteiden prenataalisen muodostumisen aikana.
Norman-Robertsin oireyhtymässä kortikaalisia gyraalisia puutoksia aiheuttavat RELN-geenin poikkeavat muutokset kromosomissa 7 johtavat solunulkoisen glykoproteiini reeliinin puutteeseen. Reeliiniä tarvitaan hermostollisten kantasolujen migraation ja sijoittumisen säätelyyn aivokuoren kehityksen aikana.[ 14 ], [ 15 ], [ 16 ]
ARX-geeni koodaa non-aristalens homeoboksiproteiinia, transkriptiotekijää, jolla on tärkeä rooli etuaivoissa ja muissa kudoksissa.[ 17 ] ARX-mutaatiota sairastavilla lapsilla on muita oireita, kuten aivojen osien puuttuminen (aivojen kurjapään ageneesis), epänormaalit sukupuolielimet ja vaikea epilepsia.[ 18 ],[ 19 ]
Useita geenejä on yhdistetty lissenkefaliaan. Näitä geenejä ovat VLDLR, ACTB, ACTG1, TUBG1, KIF5C, KIF2A ja CDK5.[ 20 ]
Sytomegalovirus (CMV) on yhdistetty lissenkefalian kehittymiseen, koska sikiön aivojen verenkierto on heikentynyt. CMV-infektion vakavuus riippuu raskausajasta. Varhainen infektio aiheuttaa todennäköisemmin lissenkefalian, koska hermosolujen migraatio tapahtuu raskauden alkuvaiheessa.[ 21 ]
Lisäksi tämän poikkeavuuden mekanismiin kuuluu hermosolujen migraation epätäydellinen tai myöhäinen lopettaminen periventrikulaarisesta generatiivisesta vyöhykkeestä aivokuoreen. Tällaisissa tapauksissa kehittyy joko epätäydellinen lissenkefalia tai pachygyria, jossa muodostuu useita leveitä uria ja kierteitä (mutta useimmat niistä puuttuvat).
Oireet Lissensefalia
Tämän patologian ensimmäiset merkit (ilman aiemmin mainittuja oireyhtymiä) eivät välttämättä ilmene heti syntymän jälkeen, vaan puolentoista tai kahden kuukauden kuluttua. Ja useimmiten havaitaan seuraavia lissenkefalian kliinisiä oireita:
- lihasten hypotonia, usein yhdistettynä spastiseen halvaukseen;
- kouristukset ja yleistyneet toonis-klooniset kohtaukset (opistotonuksen muodossa);
- vaikea kehitysvammaisuus ja kasvun hidastuminen;
- neurologisten ja motoristen toimintojen heikkeneminen.
Nielemisvaikeudet aiheuttavat vaikeuksia vauvan ruokinnassa. [ 22 ]
Vakava neuromotorinen heikentyminen ilmenee usein tetraplegiana – kaikkien raajojen halvaantumisena. Käsien, sormien tai varpaiden epämuodostumat ovat mahdollisia.
Norman-Robertsin oireyhtymässä, johon liittyy tyypin I lissenkefalia, havaitaan kallon ja kasvojen alueen poikkeavuuksia: vaikea mikrokefalia, matala otsan kaltevuus ja ulkoneva leveä nenänselkä, kaukana toisistaan sijaitsevat silmät (hyperterlorismi) ja leukojen alikehittynyt kehitys (mikrognatia). [ 23 ]
Miller-Diekerin oireyhtymälle voi olla ominaista myös epänormaalin pieni pään koko, leveä, korkea otsa ja lyhyt nenä, ohimoiden painaumat (bitemporaaliset painaumat) ja matalalle asettuneet, epämuodostuneet korvat.
Vaikealle lissenkefaaliaoireyhtymälle on ominaista mikrokefalia, silmämunien koon pieneneminen (mikroftalmia), yhdistettynä verkkokalvon dysplasiaan, obstruktiiviseen vesipäähän ja puuttuvaan tai hypoplastiseen aivokurkiaiseen.
Komplikaatiot ja seuraukset
Tämän poikkeavuuden komplikaatioista asiantuntijat mainitsevat nielemishäiriön (dysfagian) ja gastroesofageaalisen refluksin; tulenkestävän (hallitsemattoman) epilepsian; usein esiintyvät ylähengitysteiden infektiot; keuhkokuumeen (mukaan lukien krooninen aspiraatio).
Lissenkefaliaa sairastavilla imeväisillä voi olla synnynnäisiä orgaanisia sydänongelmia, kuten eteisväliseinämäaukko tai monimutkainen sydänvika syanoosineen (Fallotin tetralogia). [ 24 ]
Synnytyksen jälkeisen kasvun hidastumisen seuraukset johtavat useimmissa tapauksissa kuolemaan 24 kuukauden kuluessa syntymästä.
Diagnostiikka Lissensefalia
Diagnoosi alkaa lapsen fyysisellä tutkimuksella, vanhempien sairaushistorian sekä raskauden ja synnytyksen historian selvittämisellä.
Raskauden aikana voidaan tarvita solutonta sikiön DNA-testiä, lapsivesipunktiota tai istukkasolunäytteenottoa. [ 25 ] Lisätietoja on kohdassa – Synnynnäisten sairauksien raskausdiagnoosi
Instrumentaalista diagnostiikkaa käytetään aivorakenteiden visualisointiin ja niiden toimintojen arviointiin:
- aivojen tietokonetomografia;
- aivojen magneettikuvaus (MRI);
- aivosähkökäyrä (EEG). [ 26 ]
Raskauden aikana sikiön ultraäänitutkimuksessa 20–21 viikon jälkeen voidaan epäillä lissenkefaliaa, jos päälaen takaraivon ja kalkkariinin uurteita ei ole ja aivojen Sylvian halkeama on epämuodostunut.
Differentiaalinen diagnoosi
Differentiaalidiagnostiikka muiden synnynnäisten aivovaurioiden oireyhtymien kanssa suoritetaan.
Lissenkefaliaa on yli 20 tyyppiä, joista useimmat jakautuvat kahteen pääluokkaan: klassiseen lissenkefaliaan (tyyppi 1) ja mukulakivilissenkefaliaan (tyyppi 2). Kummallakin luokalla on samanlaisia kliinisiä ilmenemismuotoja, mutta erilaiset geneettiset mutaatiot.[ 27 ]
Tyypin I lissenkefaliassa tehdyssä aivotutkimuksessa aivokuori on neljän kerroksen sijasta, kuten normaaleilla potilailla on kuusi. Tyypin 2 lissenkefaliassa aivokuori on epäjärjestyksessä ja näyttää kyhmyiseltä tai nodulaariselta, koska aivokuori on kokonaan siirtynyt muualle aivokuoren hermosolujen klustereiden avulla, joita erottaa gliomenymaalikudos. Potilailla oli myös lihas- ja silmäpoikkeavuuksia.
- Klassinen lissenkefalia (tyyppi 1):
- LIS1: Eristetty lissenkefalia ja Miller-Diekerin oireyhtymä (kasvojen dysmorfismiin liittyvä lissenkefalia). [ 28 ]
- LISX1: DCX-geenimutaatio. LIS1-mutaatioiden aiheuttamaan lissenkefaliaan verrattuna DCX:ssä aivokuori on kuusikerroksinen neljän sijaan.
- Eristetty lissenkefalia ilman muita tunnettuja geneettisiä vikoja
- Mukulakivinen lissenkefalia (tyyppi 2):
- Walker-Warburgin oireyhtymä
- Fukuyama-oireyhtymä
- Lihasten, silmien ja aivojen sairaudet
- Muita tyyppejä ei voida sijoittaa kahteen yllä olevaan ryhmään:
- LIS2: Norman-Robertsin oireyhtymä, samanlainen kuin lissenkefalia tyyppi I tai Miller-Diekerin oireyhtymä, mutta ilman kromosomin 17 deleetiota.
- LIS3
- LISX2
Mikrolissenkefalia: Tämä on yhdistelmä normaalin aivokuoren poimuttumisen puuttumista ja epänormaalin pientä päätä. Lapsilla, joilla on normaali lissenkefalia, on syntyessään normaalin kokoinen pää. Lapsilla, joilla on pieni pään koko syntyessään, diagnosoidaan yleensä mikrolissenkefalia.
On myös tärkeää erottaa toisistaan lissenkefalia ja polymikrogyria, jotka ovat erilaisia aivojen kehityshäiriöitä.
Kuka ottaa yhteyttä?
Hoito Lissensefalia
Lissenkefalia on parantumaton orgaaninen vika, joten vain tukeva ja oireenmukainen hoito on mahdollista. [ 29 ]
Ensinnäkin tämä on antikonvulsanttien ja epilepsialääkkeiden käyttö sekä gastrostomiaputken asentaminen mahalaukkuun (jos lapsi ei pysty nielemään itsenäisesti). Hieronta on hyödyllistä.
Vaikeassa vesipäässä aivo-selkäydinneste poistetaan.
Ennaltaehkäisy
Asiantuntijat suosittelevat, että tulevat vanhemmat hakeutuvat geneettiseen neuvontaan ja että raskaana olevat naiset ilmoittautuvat ajoissa synnytyslääkäreille ja gynekologeille ja käyvät läpi kaikki suunnitellut tutkimukset.
Ennuste
Lissenkefaliaa sairastavilla lapsilla ennuste riippuu sen asteesta, mutta useimmiten lapsen henkinen kehitys ei ylitä neljän tai viiden kuukauden tasoa. Ja kaikki tämän diagnoosin saaneet lapset kärsivät vakavista psykomotorisista häiriöistä ja vaikeasti hoidettavasta epilepsiasta. [ 30 ]
NINDS:n (American National Institute of Neurological Disorders and Stroke) mukaan lissenkefaliaa sairastavien ihmisten enimmäiselinajanodote on noin 10 vuotta.