
Kaikki iLive-sisältö tarkistetaan lääketieteellisesti tai se tarkistetaan tosiasiallisen tarkkuuden varmistamiseksi.
Meillä on tiukat hankintaohjeet ja vain linkki hyvämaineisiin mediasivustoihin, akateemisiin tutkimuslaitoksiin ja mahdollisuuksien mukaan lääketieteellisesti vertaisarvioituihin tutkimuksiin. Huomaa, että suluissa ([1], [2] jne.) Olevat numerot ovat napsautettavia linkkejä näihin tutkimuksiin.
Jos sinusta tuntuu, että jokin sisältö on virheellinen, vanhentunut tai muuten kyseenalainen, valitse se ja paina Ctrl + Enter.
Waldenströmin B-soluinen lymfoplasmaattinen lymfooma
Lääketieteen asiantuntija

Viimeksi tarkistettu: 12.07.2025
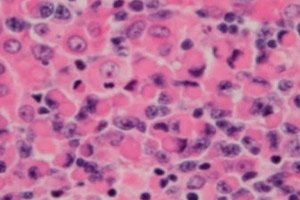
Pahanlaatuinen lymfoproliferatiivinen (immunoproliferatiivinen) sairaus, lymfoplasmasytoottinen lymfooma eli Waldenströmin makroglobulinemia, on pienten B-lymfosyyttien – imunestejärjestelmän suojaavista toiminnoista ja elimistön humoraalisesta immuniteetista vastaavien B-solujen – solukasvain. Diagnoosi tulisi tehdä vasta, kun kaikki muut pienet B-solulymfoomat on suljettu pois. Jan G. Waldenstrom kuvasi Waldenströmin makroglobulinemian vuonna 1944 ja raportoi kahdella potilaalla esiintyneistä epätavallisista lymfadenopatiaa, anemiaa, lisääntynyttä sedimentaatiota, hyperviskositeettia ja hypergammaglobulinemiaa. [ 1 ], [ 2 ]
Epidemiologia
Tämän tyyppinen lymfooma on harvinainen, hitaasti etenevä hematologinen pahanlaatuinen kasvain, ja kliiniset tilastot arvioivat sen havaitsemisasteen tässä sairausryhmässä noin 2 prosentiksi. Lisäksi miespotilaita on lähes kaksi kertaa enemmän kuin naispotilaita.
Joidenkin tietojen mukaan lymfoplasmasytoottisen lymfooman vuosittainen ilmaantuvuus Euroopan maissa on yksi 102 000 ihmistä kohden ja Yhdysvalloissa yksi 260 000 ihmistä kohden. [ 3 ]
Syyt lymfoplasmaattinen lymfooma
Useimpien onkologisten sairauksien etiologia on toistaiseksi tuntematon, mutta joidenkin sairauksien geneettisen taustan tutkimusta jatketaan. Tutkiessaan pahanlaatuisten plasmasolusairauksien, kuten B-solujen lymfoplasmasytoidisen lymfooman – Waldenströmin makroglobulinemian, syitä, tutkijat ovat löytäneet yhteyden B-lymfosyyttien patologisen lisääntymisen (solujen jakautumisen) välillä niiden erilaistumisen myöhäisessä vaiheessa tiettyjen molekyyli-geenisten häiriöiden esiintymisen kanssa, jotka muuttavat solujen perustoimintoja.
Waldenströmin makroglobulinemiaa sairastavilla potilailla on havaittu muutoksia joissakin geeneissä - somaattisia mutaatioita, eli sellaisia, jotka vaikuttavat vain kudoksiin, joissa on vaurioita erillisen klonaalisen solupopulaation geeneille ja jotka muodostavat niiden genomin variantteja, mikä johtaa syklisiin ja rakenteellisiin häiriöihin solutasolla.
Ensinnäkin nämä ovat MYD88 (L265P) -geenin ja CXCR4:n somaattisia mutaatioita, jotka koodaavat sytosolista proteiinia, jolla on tärkeä merkitys synnynnäiselle ja adaptiiviselle immuunivasteelle: adapterina se varmistaa signaalien siirtymisen tulehdusta edistävältä välittäjäaineelta IL-1:ltä (interleukiini-1) ja Toll-tyyppisiltä reseptorisoluilta, jotka aktivoivat immuunivasteen. Somaattisten mutaatioiden seurauksena tämän proteiinin molekyylin polypeptidiketjussa - sen rakenteellisessa perustassa - syntyy poikkeavuuksia. [ 4 ]
Riskitekijät
Yleisten riskitekijöiden (altistuminen kohonneille säteilytasoille, karsinogeeniset kemikaalit jne.) lisäksi seuraavia pidetään ennustajina Waldenströmin makroglobulinemian kehittymisen todennäköisyyden lisääntymiselle matala-asteisena lymfoproliferatiivisena sairautena:
- vanhuus (yli 65 vuotta);
- sukulaisten läsnäolo, joilla on tämä diagnoosi, sekä B-solujen ei-Hodgkinin lymfooma tai krooninen lymfosyyttinen leukemia;
- krooninen C-hepatiitti;
- hyvänlaatuisen monoklonaalisen gammopatian historia, idiopaattinen hematologinen sairaus, jonka ydin on lymfosyyttiplasmasolujen tuottamat epänormaalisti muuttuneet M-tyypin gammaglobuliinit;
- autoimmuunisairaudet, erityisesti Sjögrenin oireyhtymä.
Synnyssä
Joutuessaan kosketuksiin antigeenin kanssa tai T-lymfosyyttien stimuloimana jotkut B-lymfosyytit muuttuvat plasmasoluiksi – lymfosyyttisiksi plasmasoluiksi, jotka tiettyjen muutosten jälkeen alkavat tuottaa suojaavia globulaarisia proteiineja eli gammaglobuliineja (immunoglobuliineja tai vasta-aineita).
Lymfoplasmasytoidisen lymfooman/Waldenströmin makroglobulinemian patogeneesiin kuuluu B-solujen hyperproliferaatio, lymfosyyttisen plasmasolukloonin liikatuotanto ja immunoglobuliini M:n (IgM), jota kutsutaan myös monoklonaaliseksi immunoglobuliiniksi tai M-proteiiniksi, liikatuotanto veressä. Tämä on tärkein vasta-aine, jolla on suuri molekyylipaino ja pentameerinen rakenne, ja sitä tuotetaan alkuvaiheen hyökkäyksen aikana tiettyjä bakteeri- tai virusantigeenejä vastaan. [ 5 ]
Lähes kaikki tämän taudin oireet liittyvät M-proteiinin aktiivisuuden ilmentymiin, jotka voivat häiritä veren reologisia ominaisuuksia, lisätä sen viskositeettia; tunkeutua luuytimen lymfoidi- ja myeloidikudoksiin, kertyä perifeerisiin lymfoidikudoksiin (muodostuen hitaasti kasvavia kasvaimia, jotka kykenevät painostamaan ympäröiviä elimiä, hermokuituja tai verisuonia).
Vaikka krooninen lymfosyyttinen leukemia, Waldenströmin makroglobulinemia tai lymfoplasmasytoottinen lymfooma ja multippeli myelooma ovat erillisiä sairauksia, niihin kaikkiin liittyy B-lymfosyyttien lisääntynyt lisääntyminen.
Oireet lymfoplasmaattinen lymfooma
Taudin ensimmäiset oireet ovat epäspesifisiä ja niihin voi kuulua heikkous ja lisääntynyt väsymys (normokromisen anemian kehittymisen vuoksi), painonpudotus, hengenahdistus, yöllinen hyperhidroosi ja toistuva subfebriilikuume.
Lisäksi taudin alkuvaiheessa esiintyy käsien ja jalkojen herkkyyden häiriintymistä, perifeeristä neuropatiaa (jalkojen ja säärten puutumista tai pistelyä), ihon kapillaarien pieniä fokaalisia verenvuotoja (purppuraa) sekä kylmäurtikariaa (johtuen epänormaalien kryoglobuliiniproteiinien muodostumisesta ja aggregaatiosta veressä seerumissa).
Hyperviskositeettioireyhtymään liittyviä oireita ovat päänsärky ja huimaus, verkkokalvon vauriot ja näön heikkeneminen, tinnitus ja kuulon heikkeneminen, krampit, lihaskipu, korkea verenpaine, spontaanit nenäverenvuodot ja ikenien verenvuoto. Naisilla voi esiintyä kohdun verenvuotoa.
Havaittu myös: suurentuneet imusolmukkeet (lymfadenopatia); suurentunut perna (splenomegalia); sydämen vajaatoiminta, johon liittyy kardialgiaa ja sydämen rytmihäiriöitä. Vaikka viskeraalinen infiltraatio on harvinaista, maha ja suolisto voivat vaurioitua, jolloin voi kehittyä ripulia (usein rasvaisen ulosteen kera). [ 6 ], [ 7 ]
Lomakkeet
Maailman terveysjärjestön vuoden 2017 hematopoieettisten ja imukudosten kasvainten luokittelussa vahvistetaan neljä Waldenströmin makroglobulinemian diagnostista kriteeriä, mukaan lukien:
- Monoklonaalisen IgM-gammopatian esiintyminen
- Pienten lymfosyyttien tunkeutuminen luuytimeen, joka osoittaa plasmasytoidien tai plasmasolujen erilaistumista
- Luuytimen infiltraatio intertrabekulaarisella rakenteella
- Waldenströmin makroglobulinemian mukainen immunofenotyyppi, johon kuuluvat pinta-IgM+, CD19+, CD20+, CD22+, CD25+, CD27+, FMC7+, vaihteleva CD5, CD10-, CD23-, CD103- ja CD108-
Komplikaatiot ja seuraukset
Lymfoplasmasytoottista lymfoomaa sairastavilla potilailla kehittyy komplikaatioita ja seurauksia seuraavien muodossa:
- heikentynyt immuniteetti;
- luuytimen vajaatoiminta, johon liittyy sen hematopoieettisten toimintojen häiriintyminen ja anemian kehittyminen;
- tällaisten veren muodostuneiden elementtien, kuten punasolujen, leukosyyttien ja verihiutaleiden, puutos;
- ruoansulatuskanavan vauriot, joilla on krooninen ripuli ja heikentynyt suoliston imeytyminen (imeytymishäiriö);
- verisuonten seinämien tulehdus (kompleksinen immuunivaskuliitti);
- lisääntynyt luun hauraus (osteoporoosi);
- näkö- ja kuulovauriot;
- sisäelinten sekundaarinen amyloidoosi;
- eteneminen paraproteinemiseen hemoblastoosiin multippeli myelooma muodossa;
- muuttuminen erittäin pahanlaatuiseksi lymfoomaksi – diffuusiksi suurisoluiseksi B-solulymfoomaksi.
Diagnostiikka lymfoplasmaattinen lymfooma
Lymfoplasmasytoidisen lymfooman/Waldenströmin makroglobulinemian diagnosointi on yleensä vaikeaa, koska spesifisiä morfologisia, immunofenotyyppisiä tai kromosomimuutoksia ei ole. Tämä puutos tekee taudin erottamisen muista pienistä B-solulymfoomista poissulkemiskysymyksen.[ 8 ]
Lymfoplasmasytoidisen lymfooman diagnosoimiseksi tarvitaan olemassa olevien oireiden arvioinnin lisäksi yleinen ja biokemiallinen verikoe, koagulogrammi, veren proteiinien immunoelektroforeesi javeren immunoglobuliini M:n pitoisuuden määritys sekä yleinen virtsakoe. [ 9 ]
Tarvitaan luuydinbiopsia, jota varten tehdään luuydinpunktio.
Instrumentaalinen diagnostiikka suoritetaan: imusolmukkeiden ja pernan ultraäänitutkimus, luiden röntgenkuvaus, rintakehän ja vatsaontelon CT-kuvaus, oftalmoskopia.
Differentiaalinen diagnoosi
Lymfoplasmasytoottista lymfoomaa pidetään poissulkemisdiagnoosina, joten erotusdiagnoosi suoritetaan B-solujen kroonisen lymfosyyttisen leukemian, multippeli myelooman, follikulaarisen lymfooman, eri ei-Hodgkinin lymfooman alatyyppien, plasmasytooman, reaktiivisen plasmasytoosin, angiofollikulaarisen lymfoidisen hyperplasian (Castlemanin tauti) jne. kanssa.
Kuka ottaa yhteyttä?
Hoito lymfoplasmaattinen lymfooma
On pidettävä mielessä, että Waldenströmin makroglobulinemia eli lymfoplasmasytoottinen lymfooma voi olla oireeton monien vuosien ajan ja se voidaan diagnosoida M-proteiinin määrän nousuna veressä.
Jos oireita ei ole, aktiivista seurantaa suoritetaan säännöllisillä tutkimuksilla ja testeillä.
Olemassa olevien oireiden ja laboratoriokokeiden tulosten perusteella tehdään päätös hoidon aloittamisesta, joka riippuu monista tekijöistä (esim. ikä, taudin eteneminen jne.).
Protokollan mukaan tämän tyyppisen lymfooman potilaiden alkuhoito on yleensä sädehoidon ja kemoterapian yhdistelmä, johon kuuluu sytostaattien, kuten syklofosfamidin, doksorubisiinin, vinkristiinin, sekä kortikosteroidien - metprednisolonin tai deksametasonin (deksasonin) - antaminen.
Monoklonaalisten vasta-aineiden ryhmään kuuluvien lääkkeiden, erityisesti rituksimabin, kemoterapian tehokkuus on todistettu. [ 10 ]
Yleistyneissä tautitapauksissa rituksimabia käytetään yhdessä kasvainten vastaisten nukleosidianalogien (pentostatiini, kladribiini) kanssa. Hitaasti etenevässä taudissa, jossa monoklonaalisen immunoglobuliini M:n pitoisuudet ovat alhaiset, käytetään rituksimabin lisäksi sytostaattina klorambusiilia (leukeraania). [ 11 ]
Veren viskositeetin vähentämiseksi ja sen muodostuneiden elementtien tason vakauttamiseksi käytetään terapeuttista hemafereesiä.
Kun veren vasta-ainetaso on kriittisen alhainen, suoritetaan korvaushoito immunoglobuliineilla samanaikaisten toistuvien infektioiden estämiseksi.
Kuten onkohematologit toteavat, vaikka hoito voi johtaa taudin remissioon, useimmilla potilailla ilmenee uusiutuminen. Jos se tapahtuu alle 24 kuukauden kuluttua, voidaan käyttää kasvainten vastaista lääkettä, kuten ibrutinibia (tablettimuodossa). Myöhempien uusiutumisten sattuessa hoito suoritetaan alkuperäisen hoito-ohjelman mukaisesti. [ 12 ], [ 13 ], [ 14 ]
Ennaltaehkäisy
Asiantuntijat määrittävät lymfoplasmasytoidisen lymfooman ennusteen kansainvälisen ennustejärjestelmän mukaisesti arvioimalla tärkeimpiä parametreja: potilaan ikää sekä seerumin hemoglobiini-, verihiutale-, beeta-2-mikroglobuliinin ja monoklonaalisen immunoglobuliinin pitoisuuksia. [ 15 ], [ 16 ]
Tämän diagnoosin keskimääräinen eloonjäämisaste on noin viisi vuotta, mutta lähes 40 % potilaista elää kymmenen vuotta tai kauemmin.