
Kaikki iLive-sisältö tarkistetaan lääketieteellisesti tai se tarkistetaan tosiasiallisen tarkkuuden varmistamiseksi.
Meillä on tiukat hankintaohjeet ja vain linkki hyvämaineisiin mediasivustoihin, akateemisiin tutkimuslaitoksiin ja mahdollisuuksien mukaan lääketieteellisesti vertaisarvioituihin tutkimuksiin. Huomaa, että suluissa ([1], [2] jne.) Olevat numerot ovat napsautettavia linkkejä näihin tutkimuksiin.
Jos sinusta tuntuu, että jokin sisältö on virheellinen, vanhentunut tai muuten kyseenalainen, valitse se ja paina Ctrl + Enter.
Perinnöllinen nefriitti (Alportin oireyhtymä) lapsilla
Lääketieteen asiantuntija

Viimeksi tarkistettu: 05.07.2025
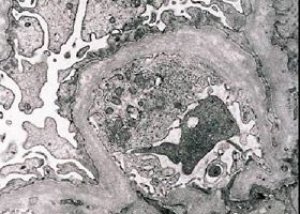
Perinnöllinen nefriitti (Alportin oireyhtymä) on geneettisesti määräytyvä perinnöllinen ei-immuuni glomerulopatia, joka ilmenee hematuriana (joskus proteinurian kanssa), munuaisten toiminnan asteittaisena heikkenemisenä kroonisen munuaisten vajaatoiminnan kehittyessä, usein yhdistettynä sensorineuraaliseen kuurouteen ja näkövammaan.
Taudin kuvasi ensimmäisen kerran vuonna 1902 L. G. Guthrie, joka havaitsi perheen, jossa hematuriaa havaittiin useissa sukupolvissa. Vuonna 1915 A. F. Hurst kuvasi uremian kehittymistä saman perheen jäsenillä. Vuonna 1927 A. Alport tunnisti ensimmäisenä kuulon heikkenemisen useilla hematuriasta kärsivillä sukulaisilla. 1950-luvulla kuvattiin silmävaurioita samankaltaisessa sairaudessa. Vuonna 1972 Hinglais ym. havaitsivat perinnöllistä hematuriaa sairastavilla potilailla munuaiskudoksen morfologisessa tutkimuksessa glomerulaaristen tyvikalvojen epätasaisen laajenemisen ja kerrostumisen. Vuonna 1985 tunnistettiin perinnöllisen nefriitin geneettinen perusta - mutaatio tyypin IV kollageenigeenissä (Fiengold ym., 1985).
Taudin geneettisen luonteen tutkimus antoi meille mahdollisuuden päätellä, että perinnöllisen nefriitin fenotyyppisten ilmentymien erot (kuulon heikkenemisen kanssa tai ilman) johtuvat mutanttigeenin ilmentymisasteesta. Näin ollen tällä hetkellä kaikkia kliinisiä variantteja pidetään yhden taudin ilmentyminä, ja termi "perinnöllinen nefriitti" on synonyymi termille "Alportin oireyhtymä".
Epidemiologisten tutkimusten mukaan perinnöllistä nefriittiä esiintyy 17 tapauksella 100 000 lasta kohden.
 [
[ Alportin oireyhtymän syyt
Taudin geneettinen perusta on mutaatio tyypin IV kollageenin a-5-ketjun geenissä. Tämä tyyppi on universaali munuaisten, simpukan, linssin kapselin, verkkokalvon ja sarveiskalvon tyvikalvoille, mikä on osoitettu tutkimuksissa, joissa on käytetty tätä kollageenifraktiota vastaan suunnattuja monoklonaalisia vasta-aineita. Viime aikoina on osoitettu mahdollisuus käyttää DNA-koettimia perinnöllisen nefriitin synnytystä edeltävässä diagnostiikassa.
Korostetaan kaikkien perheenjäsenten DNA-koettimilla testaamisen tärkeyttä mutanttigeenin kantajien tunnistamiseksi, mikä on erittäin tärkeää tätä tautia sairastavien perheiden lääketieteellisessä ja geneettisessä neuvonnassa. Jopa 20 prosentilla perheistä ei kuitenkaan ole munuaissairaudesta kärsiviä sukulaisia, mikä viittaa poikkeavan geenin spontaanien mutaatioiden korkeaan esiintyvyyteen. Useimmilla perinnöllistä nefriittiä sairastavilla potilailla on suvussa henkilöitä, joilla on munuaissairaus, kuulonalenema ja näköhäiriöitä; verisukulaisavioliitot yhden tai useamman esivanhemman kanssa ovat tärkeitä, koska sukulaisten avioliitoissa samojen geenien saamisen todennäköisyys molemmilta vanhemmilta kasvaa. On vahvistettu autosomaalisesti dominantti, autosomaalisesti resessiivinen ja dominantti, X-kromosomiin kytkeytyneet tartuntareitit.
Lapsilla erotetaan yleisimmin kolme perinnöllisen nefriittityyppiä: Alportin oireyhtymä, perinnöllinen nefriitti ilman kuulon heikkenemistä ja perinnöllinen hyvänlaatuinen hematuria.
Alportin oireyhtymä on perinnöllinen munuaistulehdus, johon liittyy kuulonalenemaa. Se perustuu munuaisten, korvan ja silmän rakenteiden glomerulaarisen tyvikalvon kollageenin rakenteen yhdistettyyn häiriöön. Klassisen Alportin oireyhtymän geeni sijaitsee X-kromosomin pitkän varren lokuksessa 21-22 q. Useimmissa tapauksissa se periytyy dominanttisti X-kromosomiin liittyneenä. Tässä suhteessa Alportin oireyhtymä on miehillä vakavampi, koska naisilla mutanttigeenin toimintaa kompensoi toisen, vahingoittumattoman kromosomin terve alleeli.
Perinnöllisen nefriitin kehittymisen geneettinen perusta on tyypin IV kollageenin alfaketjujen geenien mutaatiot. Tyypin IV kollageeni G:stä tunnetaan kuusi alfaketjua: a5- ja a6-ketjujen geenit (Col4A5 ja Col4A5) sijaitsevat X-kromosomin pitkällä haaralla 21-22q-vyöhykkeellä; a3- ja a4-ketjujen geenit (Col4A3 ja Col4A4) sijaitsevat toisessa kromosomissa; a1- ja a2-ketjujen geenit (Col4A1 ja Col4A2) sijaitsevat 13. kromosomissa.
Useimmissa tapauksissa (80–85 %) taudin periytymismalli havaitaan X-kromosomiin kytkeytyvällä tavalla, joka liittyy Col4A5-geenin vaurioitumiseen deleetion, pistemutaatioiden tai silmukointihäiriöiden seurauksena. Tällä hetkellä on löydetty yli 200 Col4A5-geenin mutaatiota, jotka ovat vastuussa tyypin IV kollageenin a5-ketjujen synteesin häiriöistä. Tämän tyyppisessä periytymisessä tauti ilmenee molemmilla sukupuolilla lapsilla, mutta pojilla se on vakavampi.
Tyypin IV kollageenin a3- ja a4-ketjujen synteesistä vastaavien Col4A3- ja Col4A4-geenien lokusten mutaatiot periytyvät autosomaalisesti. Tutkimusten mukaan autosomaalisesti dominanttia periytymistä havaitaan 16 %:lla perinnöllisistä nefriiteistä ja autosomaalisesti resessiivistä tyyppiä 6 %:lla potilaista. Col4A3- ja Col4A4-geenien mutaatioista tunnetaan noin 10 varianttia.
Mutaatioiden seurauksena tyypin IV kollageenin kokoonpanoprosessit häiriintyvät, mikä johtaa sen rakenteen häiriintymiseen. Tyypin IV kollageeni on yksi glomerulaarisen tyvikalvon, simpukan ja silmän linssin pääkomponenteista, joiden patologia havaitaan perinnöllisen nefriitin klinikassa.
Glomerulaarisen tyvikalvon osa, tyyppi IV, koostuu pääasiassa kahdesta a1-ketjusta (IV) ja yhdestä a2-ketjusta (IV), ja sisältää myös a3-, a4- ja a5-ketjuja. Useimmiten X-kromosomiin kytkeytyneessä periytymisessä Col4A5-geenin mutaatioon liittyy a3-, a4-, a5- ja a6-ketjujen puuttuminen tyypin IV kollageenin rakenteesta, ja o1- ja a2-ketjujen määrä glomerulaarisen tyvikalvossa kasvaa. Tämän ilmiön mekanismi on epäselvä, ja oletetaan, että syynä ovat mRNA:n transkription jälkeiset muutokset.
Glomerulaaristen tyvikalvojen tyypin IV kollageenin rakenteessa olevien a3-, a4- ja a5-ketjujen puuttuminen johtaa niiden ohenemiseen ja haurauteen Alportin oireyhtymän alkuvaiheessa, joka ilmenee kliinisesti useammin hematuriana (harvemmin hematuriana proteinurian tai pelkän proteinurian kera), kuulon heikkenemisenä ja lentikonuksena. Taudin eteneminen johtaa tyvikalvojen paksuuntumiseen ja läpäisevyyden heikkenemiseen taudin loppuvaiheessa, jolloin niissä lisääntyvät kollageenityypit V ja VI, mikä ilmenee proteinurian lisääntymisenä ja munuaisten toiminnan heikkenemisenä.
Perinnöllisen nefriitin taustalla olevan mutaation luonne määrää pitkälti sen fenotyyppisen ilmentymän. X-kromosomin deleetion ja samanaikaisen tyypin IV kollageenin a5- ja a6-ketjujen synteesistä vastaavien Col4A5- ja Col4A6-geenien mutaation yhteydessä Alportin oireyhtymään liittyy ruokatorven ja sukupuolielinten leiomyomatoosi. Tutkimustietojen mukaan deleetion yhteydessä olevassa Col4A5-geenin mutaatiossa havaitaan patologisen prosessin suurempi vaikeusaste, munuaisvaurion yhdistelmä munuaisten ulkopuolisten ilmentymien kanssa ja kroonisen munuaisten vajaatoiminnan varhainen kehittyminen verrattuna tämän geenin pistemutaatioon.
Morfologisesti elektronimikroskopia paljastaa glomerulaaristen tyvikalvojen (erityisesti lamina densan) ohenemisen ja kerrostumisen sekä elektronitiheiden rakeiden läsnäolon. Glomerulaariset leesiot voivat olla heterogeenisiä samalla potilaalla, minimaalisista fokaalisista mesangiaalisista leesioista glomeruloskleroosiin. Alportin oireyhtymässä glomeruliitti on aina immunonegatiivinen, mikä erottaa sen glomerulonefriitista. Tyypillisiä piirteitä ovat tubulaarisen atrofian kehittyminen, lymfohistiosyyttinen infiltraatio ja "vaahtosolujen" esiintyminen lipidisulkeuminneen - lipofageina. Taudin edetessä havaitaan glomerulaaristen tyvikalvojen paksuuntumista ja voimakasta tuhoutumista.
Immuunijärjestelmässä havaitaan tiettyjä muutoksia. Perinnöllistä nefriittiä sairastavilla potilailla on alentunut Ig A -taso ja taipumus IgM-pitoisuuden nousuun veressä. IgG-taso voi olla koholla taudin alkuvaiheessa ja laskea myöhemmissä vaiheissa. IgM- ja G-pitoisuuksien nousu on mahdollisesti eräänlainen kompensoiva reaktio IgA-puutokseen.
T-lymfosyyttijärjestelmän toiminnallinen aktiivisuus vähenee; havaitaan Ig A:n synteesistä vastaavien B-lymfosyyttien selektiivinen väheneminen, immuniteetin fagosyyttinen yhteys häiriintyy, pääasiassa neutrofiilien kemotaksian ja solunsisäisten ruoansulatusprosessien häiriintymisen vuoksi.
Kun tutkitaan munuaisbiopsiaa Alportin oireyhtymää sairastavilla potilailla, elektronimikroskopiatiedot paljastavat glomerulaarisen tyvikalvon ultrastruktuurisia muutoksia: glomerulaarisen tyvikalvon ohenemista, rakenteen häiriintymistä ja halkeilua, jolloin sen paksuus muuttuu ja muodot ovat epätasaiset. Perinnöllisen nefriitin alkuvaiheessa vika määrää glomerulaarisen tyvikalvon ohenemisen ja haurauden.
Glomerulaaristen kalvojen oheneminen on suotuisampi merkki ja yleisempi tytöillä. Vakiomuotoisempi elektronimikroskooppinen merkki perinnöllisessä nefriitissä on tyvikalvon repeäminen, ja sen vaurioitumisen vakavuus korreloi prosessin vakavuuden kanssa.
Alportin oireyhtymän oireet lapsilla
Alportin oireyhtymän ensimmäiset oireet eristetyn virtsatieoireyhtymän muodossa havaitaan useimmiten ensimmäisten kolmen elinvuoden lapsilla. Useimmissa tapauksissa tauti havaitaan sattumalta. Virtsatieoireyhtymä havaitaan lapsen ennaltaehkäisevän tutkimuksen aikana, ennen päivähoitoon ottamista tai ARVI:n aikana. Jos virtsassa havaitaan patologiaa ARVI:n aikana. Perinnöllisessä nefriitissä, toisin kuin hankitussa glomerulonefriitissä, ei ole piilevää vaihetta.
Taudin alkuvaiheessa lapsen terveys kärsii vain vähän, ja tyypillistä on virtsatieoireyhtymän pysyvyys ja vastustuskyky. Yksi tärkeimmistä oireista on vaihtelevan vaikeusasteinen hematuria, jota havaitaan 100 %:ssa tapauksista. Hematurian asteen lisääntymistä havaitaan hengitystieinfektioiden, fyysisen rasituksen tai ennaltaehkäisevien rokotusten aikana tai niiden jälkeen. Proteinuria ei useimmissa tapauksissa ylitä 1 g/vrk, taudin alussa se voi olla epävakaata, ja prosessin edetessä proteinuria lisääntyy. Virtsasedimentissä voi ajoittain esiintyä leukosyturiaa, jossa lymfosyyttejä on eniten, mikä liittyy interstitiaalisten muutosten kehittymiseen.
Myöhemmin munuaisten osittainen toiminta heikkenee ja potilaan yleistila huononee: ilmenee päihtymystä, lihasheikkoutta, valtimoiden hypotensiota, usein kuulon heikkenemistä (etenkin pojilla) ja joskus näköhäiriöitä. Päihtymys ilmenee kalpeutena, väsymyksenä ja päänsärkynä. Taudin alkuvaiheessa kuulon heikkeneminen havaitaan useimmissa tapauksissa vain audiografialla. Alportin oireyhtymässä kuulon heikkeneminen voi ilmetä lapsuuden eri vaiheissa, mutta useimmiten kuulon heikkeneminen diagnosoidaan 6–10 vuoden iässä. Lapsilla kuulon heikkeneminen alkaa korkeilla taajuuksilla, saavuttaen merkittävän asteen ilma- ja luujohtuvuudessa, siirtyen ääntä johtavasta ääntä havaitsevaan kuulon heikkenemiseen. Kuulon heikkeneminen voi olla yksi taudin ensimmäisistä oireista ja voi edeltää virtsatieoireyhtymää.
20 prosentilla Alportin oireyhtymää sairastavista potilaista havaitaan muutoksia näköelimissä. Yleisimmin havaitut poikkeavuudet ovat linssin poikkeavuuksia: sferofokia, etummainen, takimmainen tai sekamuotoinen lentikonus ja erilaiset kaihit. Alportin oireyhtymää sairastavien perheiden välillä on merkittävä mykiön esiintyvyys. Useat tutkijat havaitsevat näissä perheissä jatkuvasti molemminpuolisia perimakulaarisia muutoksia kirkkaanvalkoisten tai kellertävien rakeiden muodossa keltarauhasessa. He pitävät tätä merkkiä jatkuvana oireena, jolla on korkea diagnostinen arvo Alportin oireyhtymässä. KS Chugh ym. (1993) havaitsivat oftalmologisessa tutkimuksessa Alportin oireyhtymää sairastavilla potilailla näöntarkkuuden heikkenemistä 66,7 prosentissa tapauksista, etummaista lentikonusta 37,8 prosentissa, verkkokalvon täpliä 22,2 prosentissa, kaihia 20 prosentissa ja keratokonusta 6,7 prosentissa tapauksista.
Joillakin perinnöllistä nefriittiä sairastavilla lapsilla, erityisesti munuaisten vajaatoiminnan kehittyessä, havaitaan merkittävä viive fyysisessä kehityksessä. Munuaisten vajaatoiminnan edetessä kehittyy valtimoverenpainetauti. Lapsilla se havaitaan useammin murrosiässä ja vanhemmilla ikäryhmillä.
Perinnöllistä nefriittiä sairastaville potilaille on ominaista erilaisten (yli 5–7) sidekudosdysmorfogeneesiin liittyvien stigmien esiintyminen. Potilaiden sidekudosstigmoista yleisimpiä ovat silmien hypertelorismi, korkea kitalaki, purennan poikkeavuudet, korvalehtien epänormaali muoto, käsien pikkusormen kaarevuus ja jalkojen "sandaaliaukko". Perinnölliselle nefriitille on ominaista dysmorfogeneesistigmien yhdenmukaisuus suvun sisällä sekä niiden korkea esiintymistiheys sairauden kantajien sukulaisten keskuudessa.
Taudin alkuvaiheessa havaitaan erillinen munuaisten osittaisten toimintojen heikkeneminen: aminohappojen kuljetus, elektrolyyttien, keskittymisfunktion, asidogeneesin. Myöhemmät muutokset vaikuttavat sekä nefronin proksimaalisen että distaalisen osan toiminnalliseen tilaan, ja niille on ominaista yhdistettyjen osittaisten häiriöiden esiintyminen. Glomerulaarisen suodatuksen heikkeneminen tapahtuu myöhemmin, useammin murrosiässä. Perinnöllisen nefriitin edetessä kehittyy anemia.
Näin ollen perinnölliselle nefriitille on ominaista taudin vaiheittainen kulku: ensin piilevä vaihe tai piilevät kliiniset oireet, jotka ilmenevät minimaalisina muutoksina virtsatieoireyhtymässä, sitten prosessi asteittain dekompensoituu munuaisten toiminnan heikkenemisen ja ilmeisten kliinisten oireiden (päihtymys, astenia, kehitysviive, anemia) myötä. Kliiniset oireet ilmenevät yleensä riippumatta tulehdusreaktion kerrostuneisuudesta.
Perinnöllinen nefriitti voi ilmetä eri ikäkausina, mikä riippuu geenin toiminnasta, joka on tukahdutetussa tilassa tiettyyn aikaan asti.
Luokitus
Perinnöllistä nefriittiä on kolmea tyyppiä
- Vaihtoehto I - kliinisesti ilmenee munuaistulehduksena, johon liittyy hematuriaa, kuulon heikkenemistä ja silmävaurioita. Munuaisten vajaatoiminnan kehittyessä munuaistulehdus etenee etenevästi. Perintötyyppi on dominantti ja liittyy X-kromosomiin. Morfologisesti havaitaan tyvikalvon rakenteen häiriöitä, sen ohenemista ja halkeilua.
- Vaihtoehto II - kliinisesti ilmenee nefriitinä ja hematuriana ilman kuulon heikkenemistä. Nefriitin kulku on etenevä kroonisen munuaisten vajaatoiminnan kehittyessä. Perintötyyppi on dominantti ja liittyy X-kromosomiin. Morfologisesti havaitaan glomerulaaristen kapillaarien tyvikalvon (erityisesti laminadensan) ohenemista.
- Vaihtoehto III - hyvänlaatuinen familiaalinen hematuria. Taudin kulku on suotuisa, kroonista munuaisten vajaatoimintaa ei kehity. Perintötapa on autosomaalisesti dominantti tai autosomaalisesti peittyvä. Autosomaalisesti peittyvässä perintötavassa taudin kulku on naisilla vakavampi.
Alportin oireyhtymän diagnoosi
Seuraavia kriteerejä ehdotetaan:
- vähintään kahden nefropatiaa sairastavan potilaan läsnäolo kussakin perheessä;
- hematuria nefropatian johtavana oireena probandissa;
- kuulonaleneman esiintyminen ainakin yhdellä perheenjäsenellä;
- kroonisen munuaisten vajaatoiminnan kehittyminen yhdellä tai useammalla sukulaisella.
Erilaisten perinnöllisten ja synnynnäisten sairauksien diagnostiikassa suuri merkitys on kokonaisvaltaisella lähestymistavalla tutkimukseen ja ennen kaikkea lapsen sukupuuta koottaessa saatujen tietojen huomioimisella. Alportin oireyhtymän diagnoosi katsotaan päteväksi tapauksissa, joissa potilaalla havaitaan 3/4 tyypillisestä oireesta: hematuria ja krooninen munuaisten vajaatoiminta suvussa, neurosensorisen kuulon heikkenemisen esiintyminen, näköhäiriöt potilaalla, glomerulaarisen tyvikalvon repeämisen merkkien havaitseminen sen paksuuden muuttuessa ja epätasaiset muodot koepalan elektronimikroskooppisissa ominaisuuksissa.
Potilaan tutkimuksen tulisi sisältää kliiniset ja geneettiset tutkimusmenetelmät; sairaushistorian kohdennettu tutkimus; potilaan yleistutkimus ottaen huomioon diagnostisesti merkittävät kriteerit. Kompensaatiovaiheessa patologia voidaan havaita vain keskittymällä sellaisiin oireyhtymiin kuin perinnöllisen rasituksen esiintyminen, hypotensio, useat dysembryogeneesin stigmat ja muutokset virtsatieoireyhtymässä. Dekompensaatiovaiheessa voi esiintyä munuaisten ulkopuolisia oireita, kuten vaikea myrkytys, astenia, viivästynyt fyysinen kehitys, anemia, jotka ilmenevät ja pahenevat munuaisten toiminnan asteittaisen heikkenemisen myötä. Useimmilla potilailla munuaisten toiminnan heikkenemisen myötä havaitaan seuraavaa: asido- ja aminogeneesin heikkeneminen; 50 % potilaista havaitsee munuaisten eritystoiminnan merkittävän heikkenemisen; virtsan optisen tiheyden rajoitetut vaihtelut; suodatusrytmin häiriöt ja sitten glomerulaarisen suodatuksen heikkeneminen. Krooninen munuaisten vajaatoiminta diagnosoidaan, kun potilaiden veren seerumin ureapitoisuus on kohonnut (yli 0,35 g/l) 3–6 kuukauden ajan tai kauemmin ja glomerulaarinen suodatus on laskenut 25 prosenttiin normaalista.
Perinnöllisen nefriitin erotusdiagnostiikka tulisi suorittaa ensisijaisesti hankitun glomerulonefriitin hematuurisessa muodossa. Hankinnaisella glomerulonefriitillä on useimmiten akuutti alkamisaika 2–3 viikkoa infektion jälkeen, ekstrareunaalisia oireita, mukaan lukien hypertensio ensimmäisistä päivistä lähtien (perinnöllisessä nefriitissä päinvastoin hypotensio), glomerulusfiltraation heikkeneminen taudin alussa, eikä osittaisten tubulustoimintojen heikkenemistä ole, kun taas perinnöllisessä nefriitissä niitä on. Hankinnaisessa glomerulonefriitissä esiintyy voimakkaampaa hematuriaa ja proteinuriaa sekä kohonnutta laskoa (ESR). Diagnostisesti arvokkaita ovat perinnölliselle nefriitille tyypilliset muutokset glomerulusten tyvikalvossa.
Differentiaalidiagnostiikka dysmetabolisesta nefropatiasta suoritetaan kroonisessa munuaisten vajaatoiminnassa, perheessä on kliinisesti havaittu heterogeenisiä munuaissairauksia, ja nefropatia voi olla monenlaista pyelonefriitistä virtsakivitautiin. Lapsilla on usein valituksia vatsakivusta ja ajoittain virtsatessa, virtsan sedimentissä - oksalaateista.
Jos epäillään perinnöllistä nefriittiä, potilas tulee lähettää erikoistuneelle nefrologian osastolle diagnoosin selventämiseksi.
Mitä on tutkittava?
Kuinka tarkastella?
Mitä testejä tarvitaan?
Kuka ottaa yhteyttä?
Alportin oireyhtymän hoito
Hoitoon kuuluu rajoituksia raskaalle fyysiselle rasitukselle ja altistumiselle raittiille ilmalle. Ruokavalio on täydellinen, ja siinä on riittävästi täysipainoisia proteiineja, rasvoja ja hiilihydraatteja ottaen huomioon munuaisten toiminta. Kroonisten infektiopesäkkeiden havaitseminen ja hoito on erittäin tärkeää. Käytetään seuraavia lääkkeitä: ATP, kokarboksylaasi, pyridoksiini (enintään 50 mg/vrk), karnitiinikloridi. Kuureja annetaan 2-3 kertaa vuodessa. Hematurian hoitoon määrätään rohdosvalmisteita - nokkosta, marja-aroniamehua, siankärsämöä.
Ulkomaisessa ja kotimaisessa kirjallisuudessa on raportteja prednisolonilla hoidosta ja sytostaattien käytöstä. Vaikutusta on kuitenkin vaikea arvioida.
Kroonisessa munuaisten vajaatoiminnassa käytetään hemodialyysiä ja munuaisensiirtoa.
Perinnölliseen nefriittiin ei ole olemassa spesifistä (tehokasta patogeneettistä) hoitoa. Kaikki hoitotoimenpiteet on tarkoitettu ehkäisemään ja hidastamaan munuaisten toiminnan heikkenemistä.
Ruokavalion tulee olla tasapainoinen ja runsaskalorinen ottaen huomioon munuaisten toiminnallinen tila. Toiminnallisten häiriöiden puuttuessa lapsen ruokavalion tulisi sisältää riittävästi proteiineja, rasvoja ja hiilihydraatteja. Munuaisten vajaatoiminnan oireiden ilmetessä proteiinin, hiilihydraattien, kalsiumin ja fosforin määrää tulee rajoittaa, mikä hidastaa kroonisen munuaisten vajaatoiminnan kehittymistä.
Fyysistä aktiivisuutta tulisi rajoittaa; lapsia kehotetaan välttämään urheilua.
Tartuntatautipotilaiden kanssa kosketusta tulisi välttää ja akuuttien hengitystieinfektioiden riskiä tulisi vähentää. Kroonisten infektioiden pesäkkeiden puhdistus on välttämätöntä. Perinnöllistä nefriittiä sairastaville lapsille ei tehdä ennaltaehkäiseviä rokotuksia, rokottaminen on mahdollista vain epidemiologisista syistä.
Hormonaalinen ja immunosuppressiivinen hoito perinnöllisessä nefriitissä on tehotonta. Siklosporiini A:n ja ACE-estäjien pitkäaikaisen, usean vuoden ajan jatkuneen käytön on havaittu tuottavan joitakin positiivisia vaikutuksia (proteinurian väheneminen ja taudin etenemisen hidastuminen).
Potilaiden hoidossa käytetään lääkkeitä, jotka parantavat aineenvaihduntaa:
- pyridoksiini - 2-3 mg/kg/vrk 3 annoksessa 4 viikon ajan;
- kokarboksylaasi - 50 mg lihaksensisäisesti joka toinen päivä, yhteensä 10-15 injektiota;
- ATP - 1 ml lihaksensisäisesti joka toinen päivä, 10-15 injektiota;
- A-vitamiini - 1000 IU/vuosi/päivä yhtenä annoksena kahden viikon ajan;
- E-vitamiini - 1 mg/kg/vrk yhtenä annoksena kahden viikon ajan.
Tämän tyyppinen hoito auttaa parantamaan potilaiden yleistä tilaa, vähentämään tubulaarisia toimintahäiriöitä ja sitä suoritetaan kursseilla 3 kertaa vuodessa.
Levamisolia voidaan käyttää immunomodulaattorina - 2 mg/kg/vrk 2-3 kertaa viikossa 3-4 päivän tauoilla annosten välillä.
Tutkimustietojen mukaan hyperbaarisella hapetuksella on positiivinen vaikutus hematurian ja munuaisten vajaatoiminnan vaikeusasteeseen.
Tehokkain tapa hoitaa perinnöllistä nefriittiä on oikea-aikainen munuaisensiirto. Tässä tapauksessa tauti ei uusiudu siirron yhteydessä; pienessä osassa tapauksista (noin 5 %) siirretyssä munuaisessa voi kehittyä nefriitti, joka liittyy glomerulaarisen tyvikalvon antigeeneihin.
Lupaava suunta on synnytystä edeltävä diagnostiikka ja geenitekniikka. Eläinkokeet osoittavat, että normaalien tyypin IV kollageenin alfaketjujen synteesistä vastaavien geenien siirtäminen munuaiskudokseen on tehokasta, minkä jälkeen havaitaan normaalien kollageenirakenteiden synteesiä.
Ennuste
Perinnöllisen nefriitin ennuste on aina vakava.
Perinnöllisen nefriitin kulun ennusteellisesti epäsuotuisat kriteerit ovat:
- miespuolinen sukupuoli;
- kroonisen munuaisten vajaatoiminnan varhainen kehittyminen perheenjäsenillä;
- proteinuria (yli 1 g/vrk);
- glomerulaaristen tyvikalvojen paksuuntuminen mikroskopian mukaan;
- akustinen neuriitti;
- deleetio Col4A5-geenissä.
Hyvänlaatuisen familiaalisen hematurian ennuste on suotuisampi.